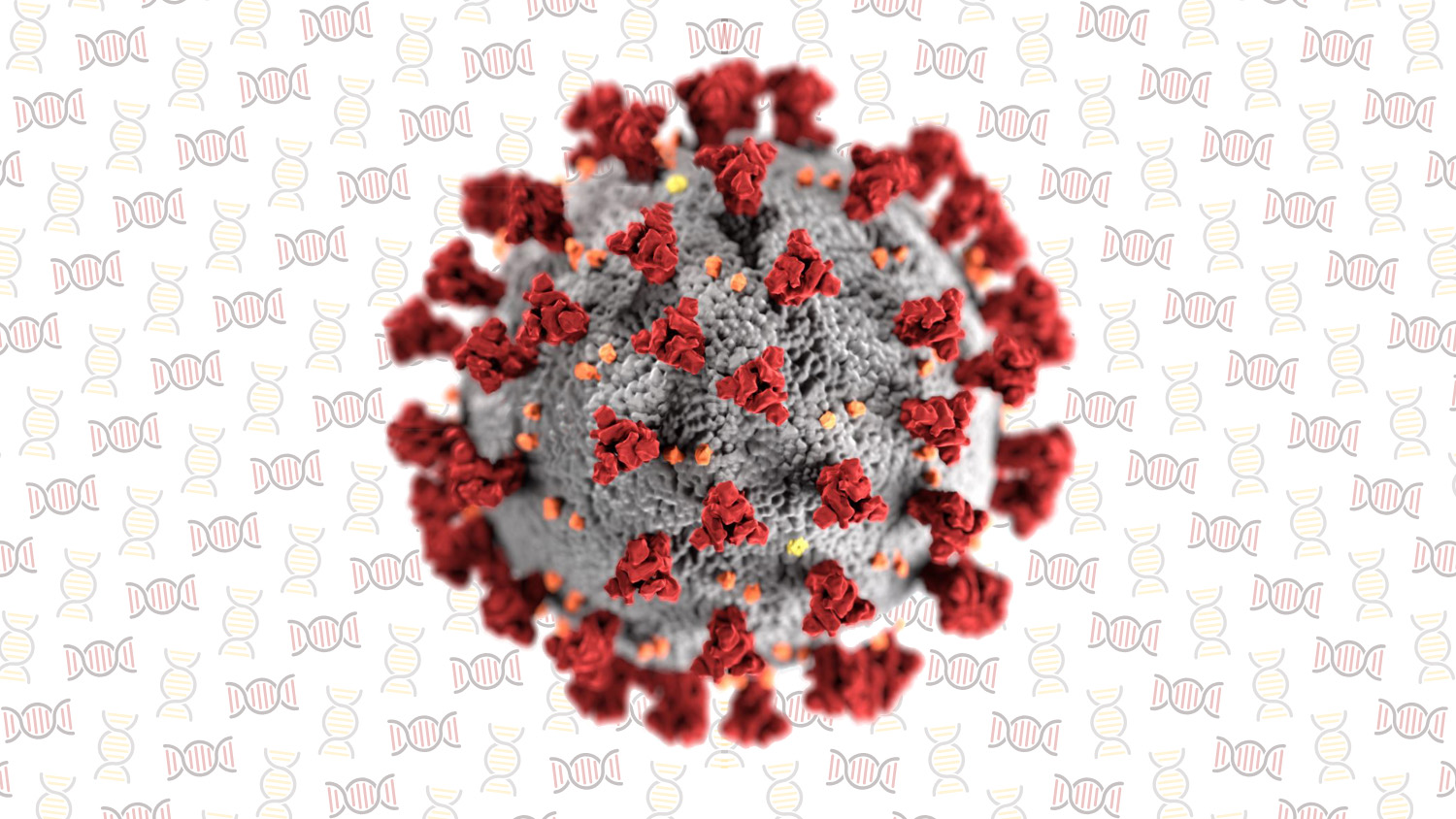
Many words have been written (or at least typed) about coronavirus SARS-CoV-2, the virus that causes Covid-19. Yet, for as much as its story has dominated news cycles and changed all of our lives, very little is being said about its biology and, specifically, its evolution. The virus is an example of the real-time consequences and dynamics of rapid evolution. It evolved, anew, in China and spread country to country, changing the world as it did. It has a power far greater than its size, a power that makes more sense in light of evolutionary biology.
To provide a little more context for current discussions of the consequences of the virus SARS-CoV-2, NC State Applied Ecology Professor (RRD) sat down with (TG) from the Center for Evolutionary Hologenomics, (KK) from Emory University, (JC), (MK), and (DR) from NC State, and (SOK) from SUNY to talk about the virus. Or rather he sat down with them, virtually, practicing good physical distancing practices.
The following conversation reflects a series of emails back and forth among the participants in response to Rob’s questions. Thanks to Olivia Sanchez Dunn for help editing the text for clarity.
Terms bold and italicized are included in the glossary at the end of the article.
The Basics
RRD: What makes a coronavirus different from any other virus?
TG: Very philosophical question, Rob! But at the most basic, there are something like 150 identified families of viruses, and the Cornaviridae are one of these families. Defined as “enveloped, positive-sense, single-stranded RNA viruses with a genome of 26-32 kilobases in length.” That is to say they have an envelope-like covering. They are based on RNA not DNA. And they are, although small compared to bacteria, large for a virus. Until recently the most famous Cornaviridae was SARS.*
Families are a key unit in the classification of viruses as they are based on and define the overall structure of a virus, e.g., what its genome is based on (DNA, RNA, single or double stranded etc), how big its genome is and what genes it contains, and what the outside of the virus looks like (enveloped, protein shell). Other famous RNA viruses from other families include HIV (within the family Retroviridae), Influenza A (within family Orthomyxoviridae), Ebola (within family Mononegavirales), Measles (within family Paramyxoviridae) and the common cold Adenoviruses (within family Adenoviridae).
RRD: How big is a coronavirus compared to, say, the simplest bacteria species? Oh, and let’s use a common set of units. Let’s measure size in terms of nucleotides (life’s genetic letters), where a kilobase (kb) is a thousand nucleotides. A megabase (Mb) is a million nucleotides.
SOK: SARS-CoV-2, the virus responsible for coronavirus disease 2019 (Covid-19), is a +ssRNA virus of approximately 30 kilobases (kb), so 30,000 nucleotides. This is big for an RNA virus. For example, flu is just 13.5 kilobases and HIV is a measly 9.5 kilobases.
TG: The genome of one of the smallest bacteria, Mycoplasma genitalium, by contrast has about 482 genes, and is about 580kb long. A bacteria species like E. coli has something like 4000-5500 genes and genome sizes of about 4.6-5.3 megabases (Mb).
RRD: So SARS-CoV-2 is 30,000 nucleotides in size, Mycoplasma genitalium is 580,000 nucleotides in size and E. coli is about 4 million nucleotides in size? That means the virus is, genetically, about a hundred times simpler than E. coli. What a streamlined little monster.
* SARS affected 26 countries and resulted in more than 8000 cases in 2003.
RRD: How many kinds of viruses are there on Earth? What proportion are unknown?
TG: Err….150 families 🙂 I imagine the answer is enormous. If you assume each species has its own unique pool of viruses…and there are millions (hundreds of millions) of unique kinds of multicellular organisms that can be hosts…that is a very big number!
RRD: And that doesn’t even include the viruses that attack bacteria, bacteriophages, right?
TG: Right.
RRD: What is a virus? Is it alive?
TG: That’s a complex question. VIruses are not alive in the same way that bacteria, plants or humans are. Viruses primarily exist as a functioning entity inside a host, whose cellular machinery is required for viruses to replicate (copy their nucleic acids, make new proteins that become new viruses, etc.). Conventional life forms-bacteria, plants and humans-use their own cellular machinery to replicate.
RRD: And the viruses also use pieces of the cells of their hosts to cloak themselves.
TG: Yup.
RRD: So they aren’t quite alive but move about the world by re-animating the bits and pieces of the bodies of their hosts? That sounds a lot like…
TG: Yes, I know. Zombies.*
* The ambiguity about whether a virus is or isn’t alive extends even to how scientists and public health officials talk about viruses. For example, a “live virus” vaccine is one in which a virus is still infective but weakened so as to be able to trigger the body’s immune response but not cause disease. In other words a live virus vaccine is made with virus that is infective and yet, as Tom points out, never fully alive. Sometimes we struggle to even have the right words to describe biological worlds distant from our perceptible world. Such is the case with viruses.
Origins and Evolution
RRD: What do we know about the origin of this SARS-CoV-2 virus that causes Covid-19? Which viruses is it most similar to? Based on what?
TG: It is a fairly close genetic relative to SARS, and the most closely related known viruses are coronaviruses found in bats. Bats are famous carriers of pathogens, as their immune system doesn’t react as strongly to pathogens as, say, ours, so infected bats often don’t have strong symptoms (as far as we can tell). There are interesting ideas on why this may be, related to the high amount of cell damage bats experience as flying animals. If their bodies over reacted to this, they’d kill themselves. So they had to evolve to tone down how they react to such damage, and this extends also to pathogens. They seem to cope fine with a belly full of viral baggage.
That bat coronaviruses are the closest relatives found so far to SARS-CoV-2 doesn’t mean it came directly from them. It more than likely jumped from bats to an intermediate host. One strong suspect being pangolins.

RRD: What might influence how able SARS-CoV-2 is to spread from human to human?
KK: Many circulating respiratory viruses in humans spread through a combination of contact transmission (two people touch each other), airborne transmission (I walk through air into which you sneezed), and environmental transmission (I touch something you touched). If a person has to be sneezed or coughed on for virus transmission to occur, this virus will probably not spread as efficiently as one that also can stay on doorknobs for 2-3 weeks, from which an individual can pick up a viral infection without ever coming near an infected individual. How well this coronavirus transmits through these different transmission routes is therefore a big factor in how able it is to spread. Another thing that would very much influence how able this virus is to spread is when an infected individual presents symptoms relative to when that individual becomes infectious. If an individual can transmit the virus before he knows he is sick, then the virus has a big advantage, and disease control efforts would become less effective.
RRD: In the news, we’ve heard that there are different “strains” of the virus. Have these new strains of the virus evolved since the origin of the virus in Wuhan, China? How do we know?
TG: A long time ago (like three days ago), it was hypothesized that there might be 2 ‘strains’, S and L. S was hypothesized to be the original strain that entered humans; L evolved from S afterwards. But more recent analyses find that the designation of these strains was premature. What looked like two strains was just variation within a single strain without any strong pattern, whether with regard to . We expect new strains to evolve, and some may have already, but the data are lacking to know for sure whether they have, much less whether any strains that do or have evolved possess new traits, such as the ability to more readily spread.
SOK: More generally, the virus will accumulate genetic mutations as it spreads among humans and expands its geographical range. Some of those mutants are either no better or worse at spreading or better at spreading and spread alongside the non-mutants. Those among them that are better at being able to reproduce, survive and spread will grow in frequency in humans.
KK: Another quick thing to add here: Some of the mutations that occur in the virus do not change the amino acids of the viral proteins and therefore are not thought to have a large effect on the fitness of the virus (that is, its transmission potential, or ability to spread between individuals). Some mutations do change the amino acids of the viral proteins, and most of the time, these mutations are deleterious. They make the virus less capable of spreading. However, a small fraction of mutations are beneficial. They increase the transmission potential of the virus.
We track viral circulation patterns by looking at all of these types of mutations. By seeing which mutations become more common, we can also start to have a better idea of which mutations may be beneficial and may be allowing the virus to adapt better to humans. Whether these mutations are indeed beneficial or not should be confirmed experimentally since other factors (just random chance, environmental factors in different geographic regions, etc.) could also impact changes in how common a mutation is in the viral population, especially early on in an epidemic.
RRD: In what other ways might the virus evolve?
SOK: Mutant strains might be better able to spread among humans, but they might also evolve the ability to spread to and among other animals. We can predict which part of the virus might be associated with this so-called “host switching.” Viruses need to attach to receptors on host cells. With SARS-CoV-2, we know that some genetic segments control how the virus attaches to the receptors of host cells and enters them. This includes, for example, something called the “receptor-binding domain” of a protein called the “spike protein,” (it is associated with the spikes on the outside of the virus). It is this genetic segment that produces this protein that recognizes the host receptor angiotensin-converting enzyme 2 (ACE2). As a result, it is the genes of this genetic segment that are important in understanding the origin of the virus.
RRD: In other words, we expect the genes associated with how a virus knocks on a cell’s door to evolve as the virus switches from hosts with one kind of door to hosts with another kind of door?
SOK: More or less, yes.
TG: Consider Influenza A as a classic example. Bird flu “jumped” into pigs where it adapted and became able to infect and spread in humans, then from pigs into things that feed on them (e.g., mink in Denmark) or are near them (humans). I’m not sure if there are documented jumps back from humans to pigs etc, but would assume so.
RRD: To say the implied here, the pig door is more similar to the human door than the bird door is to the human door, so the virus that can open the pig door stands a better chance of opening the human door.
TG: We might have extended the metaphor too far but yes, your receptors and my receptors are more like a pig’s receptors than they are like a chicken’s receptors.
MK: Backing out a little here, we primarily worry about the diseases that come from animals and start infecting people, but we give back too. . There were even of people giving their pet dogs and cats H1N1* back in 2009. And a chimp at a Chicago zoo got a different respiratory virus (human metapneumovirus) from humans. How often this happens is not known because we really don’t look for it that much.
But I’ll add that while the mutations in these viruses help them find new hosts to infect, we can also use this information to build therapies. There are some parts of the virus that have very important jobs to play, and if that protein mutates too much, or in the wrong place the virus can’t grow. When we look at the genomes of all these viruses we can see these regions and we can design drugs to work against those parts of the virus. All part of the evolutionary arms race between us and pathogens.
* H1N1, also known as “swine flu,” was responsible for the 2009 flu pandemic.

RRD: Is SARS-CoV-2 predicted to become more or less virulent as it spreads? Why?
TG: Tough question. From an evolutionary point of view…to survive the virus needs to maximise its spread and persist. If it kills too fast…it won’t get transmitted. If, however, it evolves so that it reduces its rate of replication in an individual human, there will be so little of the virus in the nasty things we spread to each other (snot, spit, other…), it won’t transmit. An “ideal” form (ideal from the perspective of the virus) would be perhaps like the common cold…it makes you feel a bit crappy, but not enough to put you in bed (thus avoiding others). It makes you sneeze and dribble, so as to pass it to others. But it doesn’t kill you which allows you to continue to sneeze, dribble and spread. Note that at this point it may already be ideal…as it doesn’t kill most people, and it certainly doesn’t kill the best people for spreading it (active young people and kids who get much more around than the elderly and infirm. So I would actually be surprised if it became much different from how it is now. There’s just not much to select on to stop it being pathogenic to the elderly/infirm.
DR: One of the reasons SARS-CoV-2 is particularly deadly is that the inflammation it causes in the lungs gives rise to pneumonia-like symptoms. This is largely self-inflicted harm arising from our own inflammatory response. It is not clear that causing such pneumonia-like symptoms in any way benefits the virus and may even reduce its transmission potential by rendering infected individuals less mobile. It’s therefore possible that there will be selection on the virus to trigger a less aggressive immune response in the lungs. But it may also be that the immune response is more under the control of the host than the virus. This certainly seems to be the case in mice where there is tremendous genetic variation among different mice strains in their immune response to SARs-like coronaviruses.
RRD: The ideal virus would make its host more social too, less willing to oblige efforts at social distancing.
TG: To continue… if it jumps from humans into yet another species alongside us (and it clearly has no problems jumping fairly large distances…even if it wasn’t directly from a bat to humans but via an intermediate…) then this could also change its rate of evolution. And possibly make it more, or less, virulent/pathogenic. To both whatever the new host species is, and new future host species, but also if it jumps back into humans.
RRD: What else can we predict about its evolution?
TG: It will certainly continue to diversify into more and more strains (or to be more precise, subclades). Whether this has any biological effect is to be seen…but one might expect it will have ramifications for the efficacy of vaccines…and mandate the development of strain specific vaccines (kind of like with influenza-A).
MK: I agree with Tom (TG), but I’ll add, it is really hard to know now what’s going to happen, but what we do, or don’t do in response will influence its evolution. Not to suggest that vaccines are bad, we need a vaccine, but as the population develops immunity that will apply new evolutionary pressures on the virus.
RRD: Which of the virus genes are most rapidly evolving? Which genes need to evolve in order to allow the virus to be successful in humans (compared to, say, bats or pangolins)?
TG: We need to talk to a virologist or someone like Eddie Holmes* who is looking at the sequences generated to date to get a good answer on this! But it’s probably genes under strongest selection to avoid the immune system or to enter cells…so on the outside of the virus.
MK: As a virologist, I’ll say that this will change over time. Early on when it was first “making the jump” to humans and evolving to spread from person-to-person there was likely pressure on lots of different genes. The outer surface genes that bind to our cells, and its other genes that are used to take over our cells. Now that it’s been in humans successfully for a while, the majority of the evolutionary pressure will be on the spike protein (again, this is the protein associated with the spikes on the outside of the virus that help it bind to host cells) and other key proteins associated with the membrane of the virus. However there will be parts of this protein that are critical for binding to the host cell’s receptor. So the evolution will likely happen in ways to conserve that part of the protein, but still change the rest in ways that our immune system doesn’t recognize it any more.
* Tom emailed Eddie. Eddie did not say which genes might be evolving fastest but did say that there was no evidence that the virulence genes yet were.
RRD: How would the evolution or spread of the virus be different if most people were vaccinated against it (imagining the existence and release of a vaccine)?
KK: There are two possible general outcomes of vaccination against a virus such as this… If vaccination rates are high enough then the virus would not be able to continue to circulate and go extinct. The level of vaccination required is called the vaccination threshold, and it depends on the transmission potential (“basic reproduction number,” R0) of the virus. The higher the transmission potential the larger proportion of the public that needs to be vaccinated to stop (or even slow) the virus. If vaccination still occurred at relatively high levels, but not sufficiently high to keep the virus from spreading, then vaccination of some individuals (provided that the vaccine is a high-efficacy, protective vaccine) would actually protect some of the unvaccinated individuals too. This is called ‘herd immunity’ and it occurs because vaccination reduces the amount of virus circulating, and therefore decreases the rate at which unvaccinated individuals also become infected.
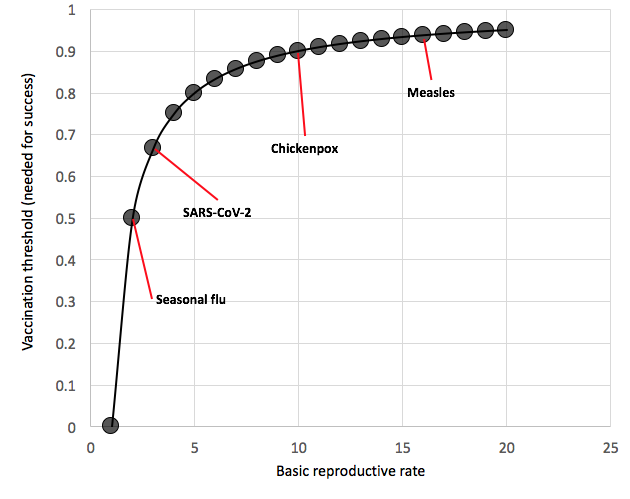
SOK: I can imagine the virus circulating in pockets of unvaccinated individuals, whether they are cases or asymptomatic.
KK: There is also a possibility that vaccination at high levels (but below the vaccination threshold) could put selection pressures on the virus to escape vaccine-provided host immunity. In this case, the vaccine may ultimately need to be reformulated to be based on a different virus strain.
DR: Human antibodies against coronaviruses target the Spike domain (the red triangles in figure 4), the same protein that the virus uses to bind human cellular receptors like ACE2 (those doors again). This puts the virus in a bit of an evolutionary predicament because if its spikes change too much they can’t bind to the host receptors, but if they don’t change enough they are attacked by the host immune system. But this predicament is not unique to coronaviruses. For example the hemagglutinin protein of influenza also serves as a receptor binding protein and is likewise a major target of human antibodies. We know from studying viruses like influenza that these proteins are fairly flexible in their ability to tolerate mutations that allow them to escape immunity while conserving their other biological functions. My prediction, although speculative, is therefore that SARS-Cov-2 will be able to escape human antibodies generated by either a vaccine or from natural immunity. Indeed, there is some preliminary evidence that other common human coronaviruses like HCoV-OC43 evolve to escape antibody-based immunity. It’s important to note that this in no way diminishes the importance of developing a vaccine, but rather that the vaccine may need to be updated overtime as Katia (KK) alluded to above.
MK: This depends on what kind of immunity is conferred, and how long it lasts. There are vaccines that provide “sterilizing immunity” and then there are others that just prevent disease. These different types of protection drive virus evolution differently. Sterilizing immunity is what we strive for under most circumstances and it’s the type of immunity the flu vaccine hopes to confer, and does when the vaccine is matched right with the circulating strain. In this case you have induced high levels of antibodies in the person before they are infected, and then when the virus shows up the antibodies block the virus from even starting. This is why every decade or so you see a shift in the dominant subtype of HA in human seasonal flu cases. When the whole population has sterilizing immunity (from vaccination or infection) that subtype doesn’t circulate very well any more and is replaced by another.
If you don’t get sterilizing immunity you end up with some low level infection (and antibodies slow but don’t stop the virus in your body), but it isn’t bad enough to send you to the doctor. This subclinical infection allows for the virus to replicate in humans and pick up new mutations and then it’s just a matter of Darwinian selection until a virus that spreads faster, or kills more quickly emerges. There are some examples of this in humans, but the best studied example is the Marek’s Disease Virus vaccine in chickens. Ever since this vaccine was first introduced in the 1950s a more virulent version of the virus has emerged every 10-20 years requiring a new vaccine.
RRD: What should we be paying attention to with regard to the biology of the virus? What are the unknowns?
TG: Will it/strains of it mutate in ways that will negate the efficacy of the diagnostic tests? Or affect the efficacy of vaccines?
RRD: Oh jeez, I hadn’t thought about the possibility that strains could evolve in such a way as to become undetectable.
MK: It is still early, but for me the big question is what does immunity to these viruses look like. How long does it last? There are several coronaviruses we all see every year. They cause the common cold. Each year we get infected by more or less the same viruses because our immunity to these viruses only lasts about 6 months. Will it be the same for SARS-CoV-2? Or maybe we’ll still get infected by SARS-CoV-2 but the next time it will just be cold.
RRD: What did I not ask that I should have (and if you pose the question, also offer something of an answer)?
TG: Covid-19 is one thing…but it is hardly going to be the only time this happens. How do we stop this happening again? And how do we deal with the slightly awkward issues that when governments act poorly (e.g. suppress the truth…downplay the effect…play on conspiracies)…it’s damned hard to stop this happening again. To be honest I think we’ve been super lucky with Covid-19 compared to what we could have ended up with.
JC: I think we need to think about how we tailor our responses to the biology of viruses. Up until Ebola, funding to state and locals was categorical by disease. We fought very hard to say all responses are, at core, the same with nuances specific to the disease and largely related to scope, scale and mechanism of transmission. Each response really did build on the previous one. Fortunately/unfortunately the outbreaks were never close enough to test if what we did was correct and many times we had to re-learn some of the practices. Additionally, there have never been strong research methods developed to test the response components, most of which were adopted from “daily” small scale public health practice or other non-health emergency response practice.
This means we made assumptions about outcomes and were surprised when, for example, H1N1 had a particularly high case fatality rate in pregnant women and the elderly were somewhat protected. This is very different from seasonal influenza which was the model for predicting populations at risk. And this wasn’t realized until well into the pandemic. Methods such as randomized control trials just don’t work during an emergency and the adaptation of other research methodologies including qualitative analysis isn’t quite respected yet.
JC: Also… For Covid-19 it would have been nice to have the research/evaluation infrastructure in place to evaluate just how quarantine was or wasn’t effective and what were the nuances that made it so.Knowing this may have made implementing layered quarantine more effective as it spread geographically. For example, community quarantine in Wuhan managed to mitigate spread outward (note that quarantine in this setting is not meant to eliminate spread, just blunt it so that others can get ready and the health care system isn’t overwhelmed ) but the cruise ship quarantine most likely failed for those on the ship. One size doesn;t fit all.
RRD: Is there a particular ability of some virus or a story about a virus (other than coronavirus) that especially fascinates you?
DR: We tend to think of most viruses as specialists that are highly adapted to infect a single species. Such specialists face tradeoffs that limit the ability of any particular strain of virus to infect multiple different species. What’s
unfortunate for us is that viruses such as SARs-like coronaviruses are actually more like generalists and can thrive in many different hosts. All they need is a little time to adapt to a new host. To me, it’s kind of inspiring that something so small and limited in terms of its genome size can be so flexible.
TG: Well there’s the giant mimiviruses which are causing some philosophical headaches about what defines a virus. Typically they are both as large in genome size and physical size as bacteria such as Rickettsia, and their genomes code for products that haven’t normally been seen in viruses, including for nucleotide and amino acid synthesis. But on the other hand they don’t have the mechanism for protein translation and energy metabolism, which are traits we normally associate with living organisms.
SOK: Influenza A virus (antigenic shift and drift). Koala retrovirus (both exogenous, i.e. transmitted between individuals, and endogenous, i.e inherited from parent to offspring).
JC: As far as what is my favorite agent from a preparedness perspective: it has to be flu. That little bugger is just too adaptive for us to get a handle on and requires a whole new approach. We have to have plans in place and a vaccine that doesn’t need annual revisions. This is all consistent with building resilience whether it’s biological, social or behavioral: absorbing the trauma and being ready for the next time.
TG: And then there are human viruses that must have an animal origin but it’s really not obvious from which animal. I think, for example, that smallpox still doesn’t have a good animal origin explanation. And there is wild ass herpes virus. Which just makes me laugh at its name.
MK: How much time do you have, said the virologist. All of the above, but probably one of my favorite topics is the relatively new field looking at horizontal gene transfer by viruses (from viruses to their hosts and hosts to viruses) and how they may have helped plants and animals make major leaps in evolution. As one example there’s some evidence that retroviruses genes actually provided the building block to .
Who Are We?
TG is Tom Gilbert: Tom studies hologenomics, namely how microbes and their hosts work as one, at the University of Copenhagen’s GLOBE Institute.
KK is Katia Koelle: Katia studies the evolution and spread of viruses, with a special focus on influenza. She is based at Emory University.
DR is David Rasmussen: David develops computational methods for studying infectious disease epidemiology and viral evolution. He is based at North Carolina State University.
SOK is Sergios-Orestis Kolokotronis: Sergios studies the molecular evolution and ecology of infectious disease systems. He is based at SUNY Downstate Health Sciences University in Brooklyn and the American Museum of Natural History in New York.
RRD is Rob Dunn: Rob studies the biology of species associated with humans. He is based in the Department of Applied Ecology at North Carolina State University and the Center for Evolutionary Hologenomics at the University of Copenhagen.
JC is Julie Casani: Julie has experience leading statewide (Maryland and North Carolina, USA) response to public health emergencies. She teaches global health at NC State University
MK is Matt Koci: Matt is a virologist and immunologist working on host-microbe interactions in birds. He is based at NC State University.





