Tohoku University researchers have created a reliable means of predicting the performance of a new and promising type of catalyst. Their breakthrough will speed up the development of efficient catalysts for both alkaline and acidic environments, thereby saving time and effort in future endeavors to create better fuel cells.
Details of their research were published in the journal Chemical Science on March 15, 2024.
Fuel cell technology has long been heralded as a promising avenue for clean energy, but challenges in catalyst efficiency have hindered its widespread adoption.
Molecular metal-nitrogen-carbon (M-N-C) catalysts boast distinctive structural properties and excellent electrocatalytic performance, particularly for the oxygen reduction reaction (ORR) in fuel cells. They offer a cost-effective alternative to platinum based catalysts.
One such variant of M-N-C catalysts are metal-doped azaphthalocyanine (AzPc). These possess unique structural properties, characterized by long stretching functional groups. When these catalysts are placed on a carbon substrate, they take on three-dimensional shapes, much like a dancer placed onto a stage. This shape change influences how well they work for ORR at different pH levels.
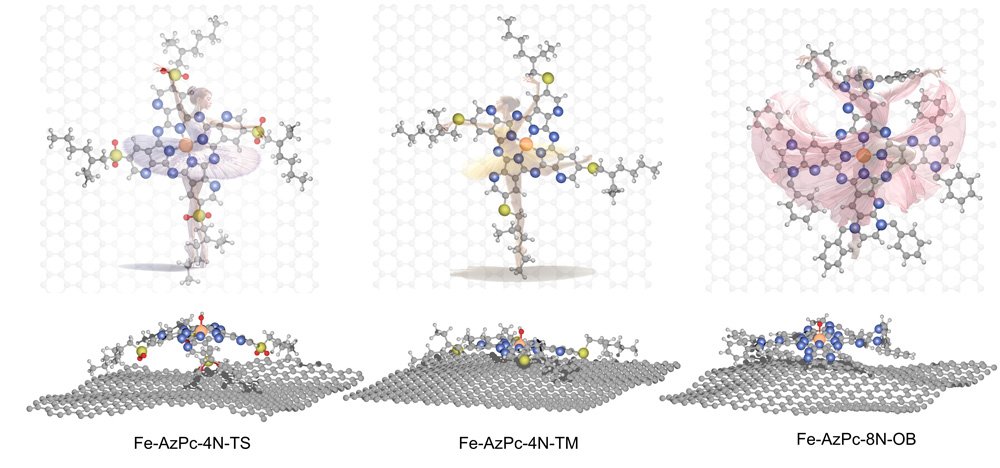
Still, translating these beneficial structural properties into increased performances is a challenge, one that requires significant modeling, validation, and experimentation, which is resource intensive.
“To overcome this, we used computer simulations to study how the performance of carbon-supported Fe-AzPcs catalyst for oxygen reduction reactions changes with different pH levels, by looking at how electric fields interact with the pH and the surrounding functional group,” says Hao Li, associate professor at Tohoku University’s Advanced Institute for Materials Research (WPI-AIMR) and corresponding author of the paper.
In analyzing Fe-AzPcs performance in ORR, Li and his colleagues incorporated large molecular structures with complex long-chain arrangements, or ‘dancing patterns,’ with arrangements of over 650 atoms.
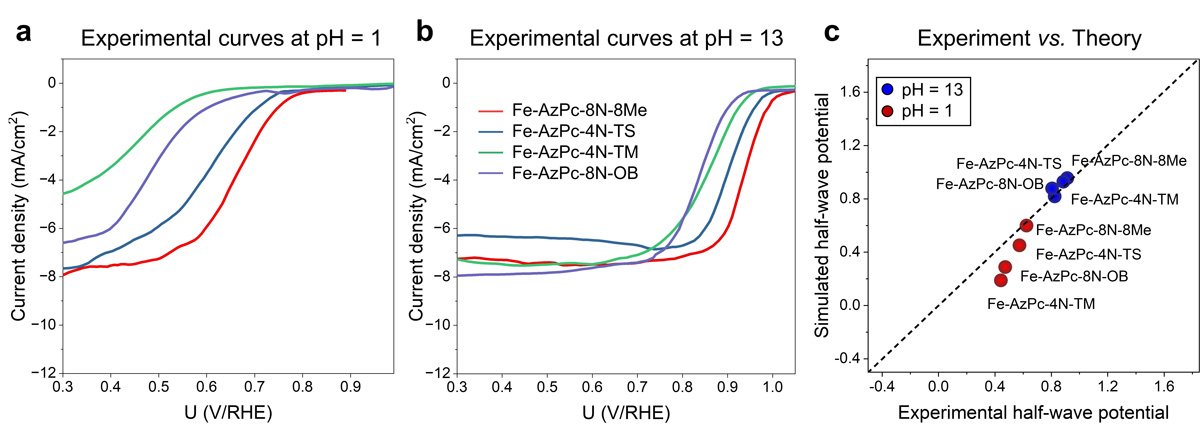
Crucially, the experimental data revealed that the pH-field coupled microkinetic modeling closely matched the observed ORR efficiency.
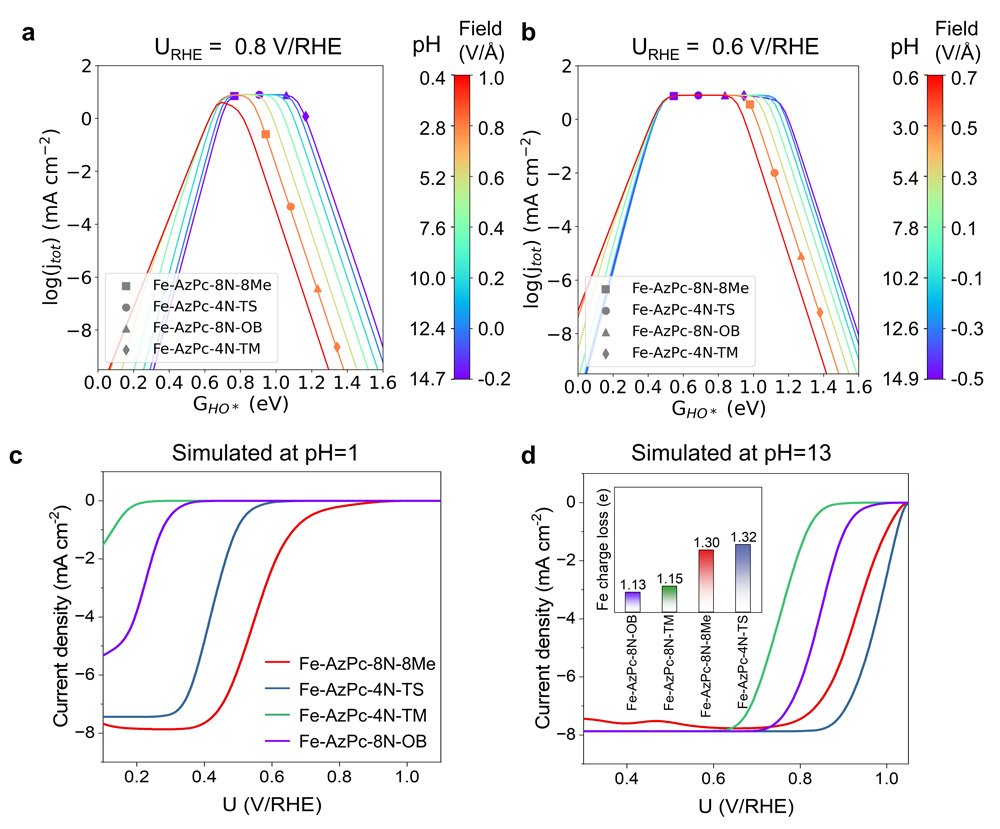
“Our findings suggest that evaluating the charge transfer occurring at the Fe-site, where the Fe atom usually loses approximately 1.3 electrons, could serve as a useful method for identifying suitable surrounding functional groups for ORR,” adds Li. “We have essentially created a direct benchmark analysis for the microkinetic model to identify effective M-N-C catalysts for ORR across various pH conditions.”
- Publication Details:
Title: Benchmarking pH-Field Coupled Microkinetic Modeling Against OxygenReduction in Large-Scale Fe-Azaphthalocyanine Catalysts
Authors: Di Zhang, Yutaro Hirai, Koki Nakamura, Koju Ito, Yasutaka Matsuo, Kosuke Ishibashi, Yusuke Hashimoto, Hiroshi Yabu, and Hao Li
Journal: Chemical Science
DOI:





